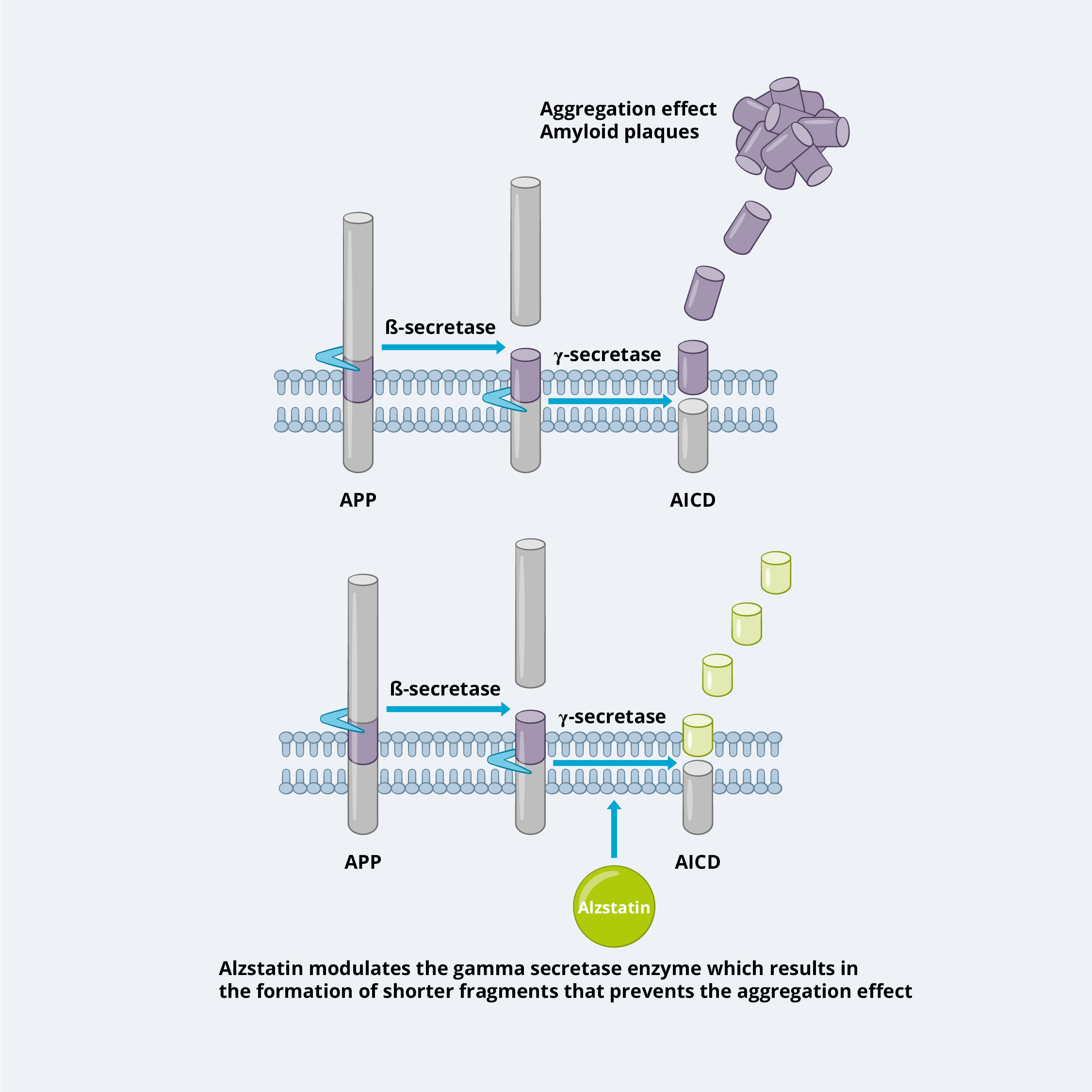
Our disease-modifying research platform Alzstatin consists of gamma-secretase modulators (GSMs). GSMs reduces the production of toxic amyloid beta (Aβ42) in the brain. Aβ42 plays a key pathogenic role in Alzheimer’s disease and begins to accumulate in the brain years before clear symptoms start to develop.
Amyloid-beta
The brain consists of about 100 billion nerve cells (neurons) that are interconnected in an intricate network and are vital for brain function and survival. Autopsies of the brains of Alzheimer’s patients show abundant amounts of amyloid plaques, the accumulation of which is assessed to have a major impact on the course of disease. Amyloid plaques consist of an accumulation of amyloid-beta (Aβ) peptides, which are formed and secreted by nerve cells in the brain.
Aβ is a family consisting of 30–43 amino acids (Aβ30 – Aβ43) and of these different forms, Aβ42 is the main component in Aβ plaques. Aβ42 is particularly “sticky” and has a strong tendency to form aggregates. This process is complex and the Aβ42 peptide accumulates in smaller aggregates, oligomers and protofibrils, which then form the building blocks of fibrils that eventually form amyloid plaques.
Aβ is formed by the sequential cleavage of the amyloid precursor protein (APP) by two enzymes, beta secretase and gamma secretase. The latter enzyme is responsible for the final cleavage into smaller fragments, including Aβ42, which over time accumulates and forms the characteristic amyloid plaques in the brains of Alzheimer’s patients.
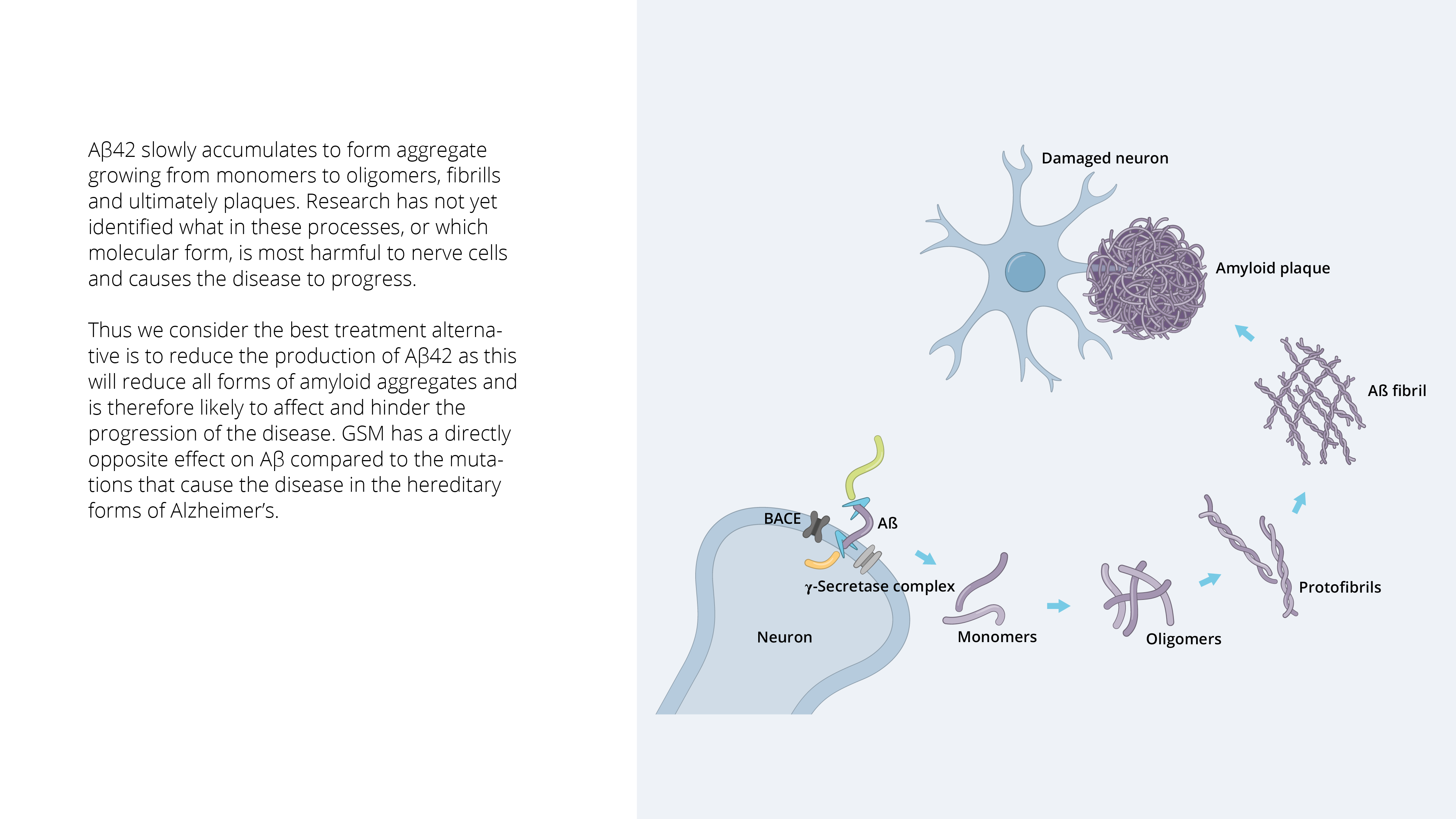
In Alzheimer’s disease, the nerve cells are surrounded by these Aβ aggregates, which affects the communication ability and function of the nerve cells, which in turn leads to them withering and eventually dying. Exactly how Aβ causes nerve cells to die at the molecular level is not yet known. Much of the data suggest that the ill health of the nerve cells leads to accumulations of another protein, tau, inside the cells and that taken together, this leads to the death of the cells. A clear hereditary connection can be seen in about 1 percent of all Alzheimer’s cases.
The heredity component involves specific mutations in any of three specific genes, all of which are directly involved in Aβ peptide production. In fact, mutations in the gamma secretase complex are one of the most common causes of hereditary Alzheimer’s disease. The common denominator among all of these disease-causing mutations is that they affect the Aβ peptide itself, or its production (relatively more Aβ42), in a way that accelerates build-up of amyloid plaques, thereby demonstrating the central role that Aβ plays in Alzheimer’s, while making this peptide and the amyloid process the most validated disease process in Alzheimer’s today. Major advances in research during the 2000s have made it possible to follow the amyloid process in living individuals over time. A large number of such studies have shown that amyloid plaques begin to accumulate up to 20 years before symptoms appear and that it more or less reaches its peak and decreases in further growth once the symptoms of the disease begin to become apparent.
When clinical symptoms occur, the structure of the brain has begun to change because of diseased nerve cells that have withered and nerve cells that have died. The brain has literally begun to decrease in size. Several previous clinical trials with Aβ-targeted drugs in patients with relatively advanced Alzheimer’s have failed. Given the new knowledge about how early Aβ builds up and is stored in the brain, it is likely that these candidates were tested too late in the disease, during a phase when Aβ had already played most of its pathogenic role. New clinical studies in the field, in which Aβ-targeted drugs have been administered earlier in the course of disease, have been able to demonstrate clinical efficacy in patients, thereby strengthening the validity of this target mechanism. It is clear that Aβ-amyloidosis is a causative agent of hereditary familial Alzheimer’s disease, as described above. More and more comparative studies, where the Aβ process in sporadic Alzheimer’s has been compared with familial Alzheimer’s, show a similar structure of Aβ in sporadic disease, though it usually occurs later in life. These research data strongly suggest that Aβ accumulation also plays a crucial pathological role in sporadic Alzheimer’s, which accounts for about 99 percent of cases in Alzheimer’s disease.
Given it key role in the production of toxic Aβ, the gamma secretase complex is a highly interesting therapeutic target for Alzheimer’s disease, and different strategies to interact with this enzyme complex have been investigated:
Gamma Secretase Inhibitors (GSIs):
GSIs are drugs that directly inhibit the activity of the gamma secretase enzyme complex. By inhibiting gamma secretase, GSIs prevent the formation of Aβ42. However, gamma secretase has other important functions in the body, including processing other proteins involved in vital cellular processes. Therefore, broad-spectrum GSIs inhibit the cleavage of multiple proteins, leading to potential side effects. The inhibition of gamma secretase can cause problems in the processing of e.g. Notch protein, which is crucial for normal cell development and differentiation. Therefore, GSIs have faced major challenges in clinical development due to their adverse effects.
Gamma Secretase Modulators (GSMs):
GSMs, on the other hand, do not directly inhibit the activity of gamma secretase but modulate its function in a more selective manner. They specifically target the cleavage of APP by gamma secretase without affecting other substrates, such as Notch. GSMs promote a shift in gamma secretase cleavage preference, leading to a decrease in the production of longer, aggregation-prone forms of Aβ (such as Aβ42) and an increase in shorter forms, such as Aβ37 and 38, which are not aggregation-prone and have been suggested to have protective effects.
The advantage of GSMs over GSIs is their relative selectivity, which reduces the risk of side effects associated with inhibiting gamma secretase activity broadly. By shifting the gamma secretase cleavage preference towards shorter forms of Aβ, GSMs aim to reduce the formation of amyloid plaques while preserving the normal physiological function of gamma secretase.
The drug candidate in the Alzstatin platform is a GSM and have shown reduction of more than 50 percent of toxic Aβ42 in vivo, without affecting other signaling pathways important for cells. Moreover, the increased production of shorter forms of the Aβ peptide, Aβ37 and Aβ38, which in addition to their not being sticky and not forming aggregates, may also have a restrictive effect on the formation of Aβ42 aggregates. Preclinical and clinical data also show that they can have protective effects. This means the drug candidate in the Alzstatin platform have two separate but synergistic effects that together contribute to a stronger anti-amyloidogenic – and thus more potent – disease-modifying effect.
Furthermore, the project is supported by positive findings made in recently published clinical patient studies with antibodies such as Aducanumab, Donanemab, and Lecanemab/BAN2401 which we believe validates the amyloid hypothesis as a treatable and clinically relevant pathological mechanism. At the CTAD conference in October 2023, Roche also presented clinical phase I data with its GSM, and was able to demonstrate PoM in humans as well as a good safety profile for this class of substances. They now have entered Phase II studies in 2024, further validating this targeting mechanism and helping to pave a regulatory path forward for this substance class. Great progress has also been made in the diagnostic field with new blood-based tests, which brings a cost-effective opportunity to screen high-risk populations, and thus identify the right patients in the presymptomatic phase of the disease for upcoming clinical studies and future treatments.
ACD680 the drug candidate within Alzstatin, is currently in the preclinical development phase. AlzeCure also sees the benefits arising from a small molecule drug, which enables oral administration (tablets), low production costs and good penetration of the blood-brain barrier.
Find our Alzstatin publications: HERE
For more information and material: see here
The video below briefly shows how a compound from the Alzstatin program acts in the brain.