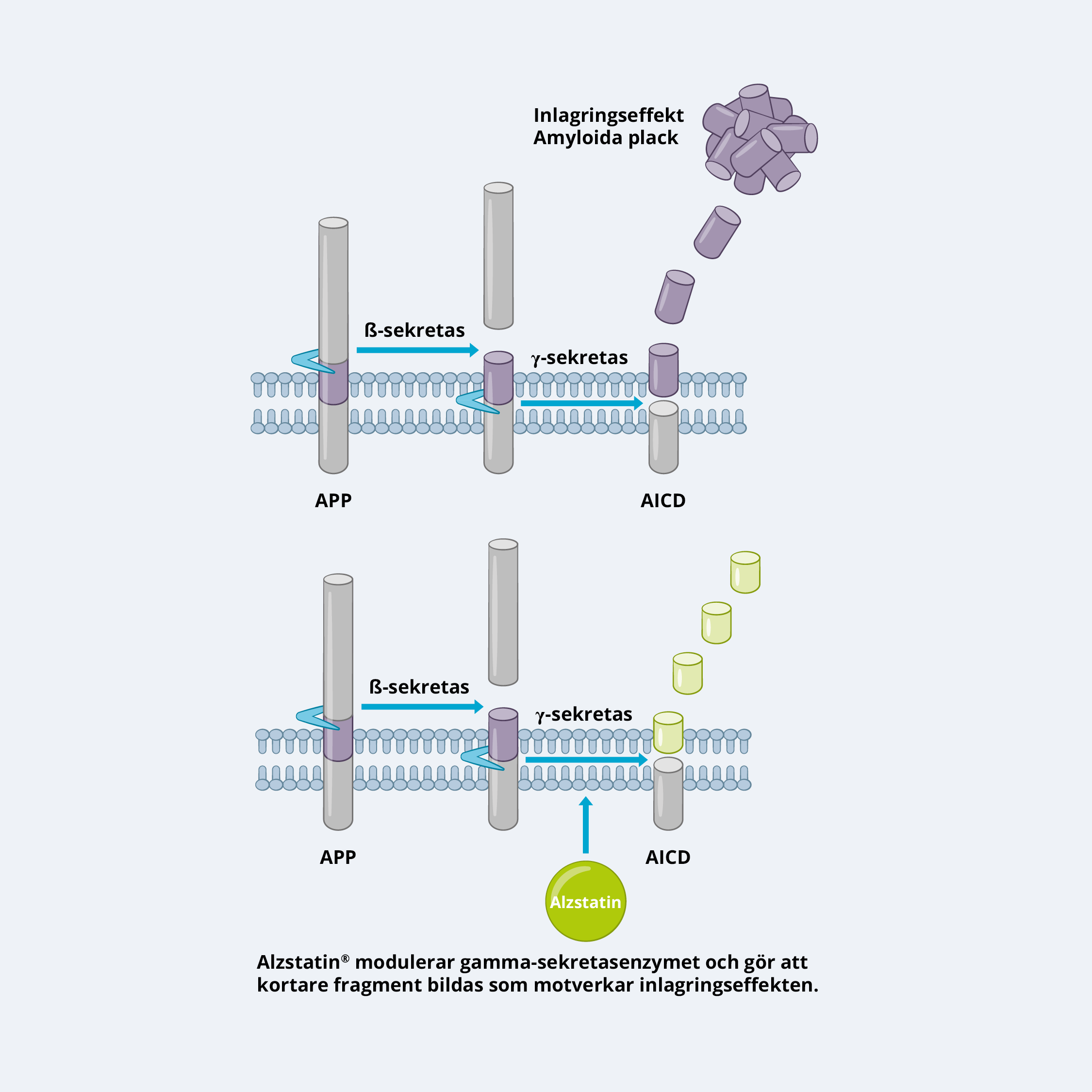
Vår sjukdomsmodifierande läkemedelsplattform Alzstatin består av gamma-sekretas modulatorer (GSM). GSM minskar produktionen av toxiskt amyloid-beta (Aβ42) i hjärnan. Aβ42 spelar en central sjukdomsframkallande roll i Alzheimers sjukdom och börjar ansamlas i hjärnan många år innan tydliga symptom börjar utvecklas.
Amyloid-beta
Hjärnan består av cirka 100 miljarder nervceller som är sammankopplade i ett intrikat nätverk och är vitala för hjärnans funktion och överlevnad. Vid obduktioner av Alzheimerpatienters hjärnor påvisas rikliga mängder av så kallade amyloida plack, vars ansamling bedöms ha en stor påverkan på sjukdomsförloppet. Amyloida plack består av en ansamling av Aβ-peptider, som bildas och utsöndras av nervceller i hjärnan.
Aβ är en familj som består av 30–43 aminosyror (Aβ30–Aβ43). Av dessa återfinns främst Aβ42 i Aβ-plack. Aβ42 är särskilt ”klibbig” och har en stark benägenhet att klumpa ihop sig. Denna process är komplex och Aβ-peptiden ansamlas i mindre aggregat, oligomerer och protofibriller, som sedan utgör byggstenar för fibriller som så småningom bildar amyloida plack.
Aβ bildas genom sekventiell klyvning av amyloid prekursorproteinet (APP) av två enzymer, beta-sekretas och gamma-sekretas. Det senare enzymet är ansvarigt för den slutliga klyvningen till mindre fragment, inklusive Aβ42, som med tiden ackumuleras och bildar de karakteristiska amyloida placken i hjärnan hos Alzheimer patienter.
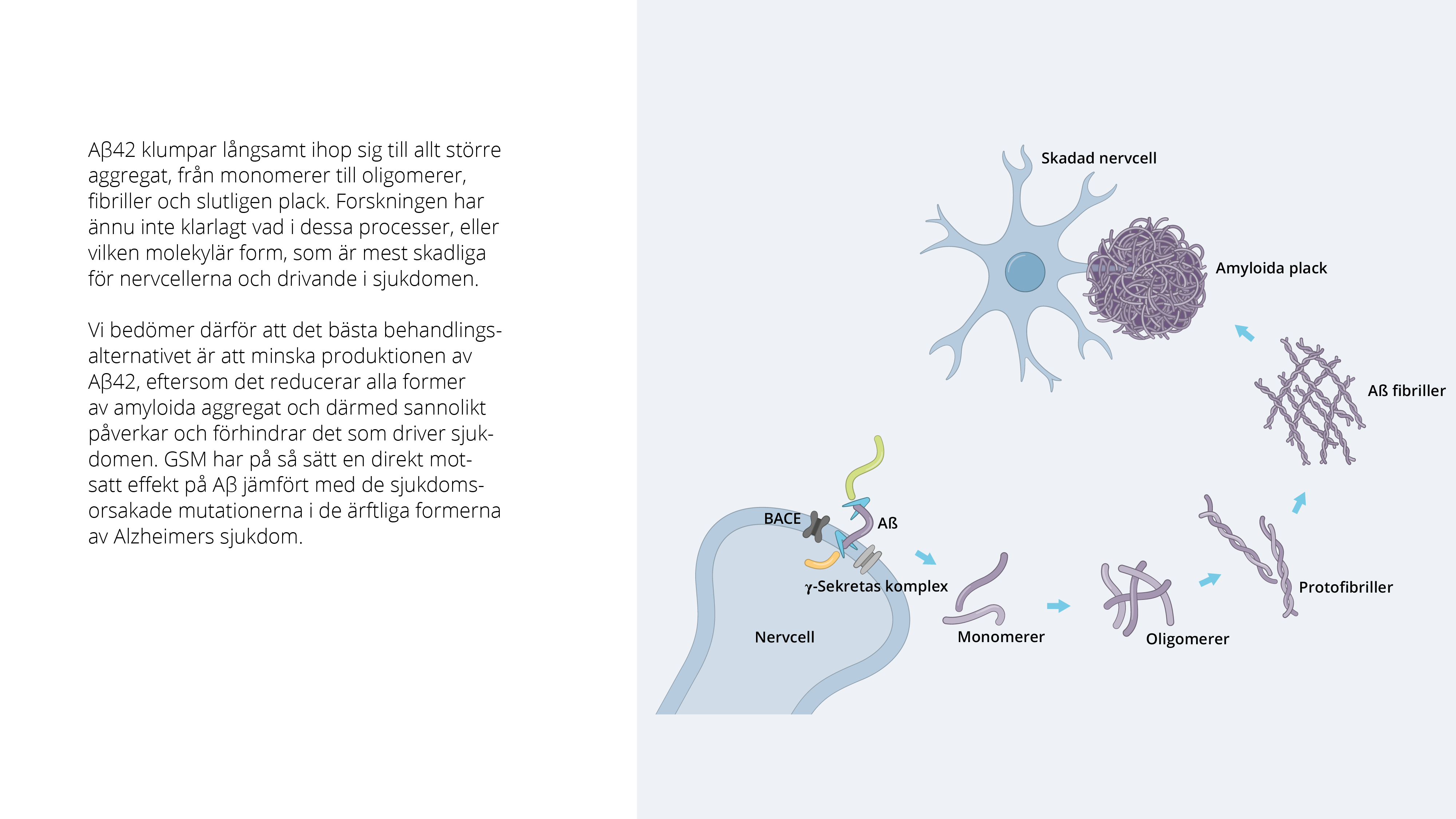
Vid Alzheimers sjukdom omges nervcellerna av dessa Aβ-aggregat, vilket påverkar nervcellernas kommunikationsförmåga och funktion, vilket i sin tur leder till att de förtvinar och slutligen dör. Exakt hur Aβ orsakar nervcellernas död på molekylär nivå är ännu inte känt. Mycket data tyder på att nervcellernas ohälsa leder till ansamlingar av ett annat protein, tau, inuti cellerna och att det sammantaget leder till att cellerna dör. I ca 1 procent av alla Alzheimerfall kan man se ett tydligt ärftligt samband. Ärftligheten består av specifika mutationer i någon av tre specifika gener som alla är direkt involverade i Aβ-produktion. Faktum är att mutationer i gammasekretas komplexet är en av de vanligaste orsakerna till ärftlig Alzheimers sjukdom. Den gemensamma nämnaren bland alla dessa mutationer är att de påverkar själva Aβ-peptiden eller dess produktion (relativt mer Aβ42) på ett sätt som resulterar i en accelererad uppbyggnad av amyloida plack.
Detta visar vilken central roll Aβ spelar i Alzheimers och gör denna peptid och den amyloida processen till den idag mest validerade sjukdomsprocessen i Alzheimers. Stora framsteg inom forskningen under 2000-talet har gjort det möjligt att följa den amyloid processen hos levande individer över tid. Ett stort antal sådana studier har visat att amyloida plack börjar ansamlas upp till 20 år innan symtomen uppträder och att den mer eller mindre når sin topp och avtar i vidare tillväxt när symptomen av sjukdomen väl börjar blir tydliga.
När kliniska symtom uppstår har hjärnans struktur börjat förändras på grund av sjuka nervceller som har dragit ihop sig och nervceller som har dött. Hjärnan har bokstavligen börjat minska i storlek. Flera tidigare kliniska prövningar med Aβ-riktade läkemedel på patienter med relativt långt utvecklad Alzheimer har misslyckats. Med tanke på den nya kunskapen om hur tidigt Aβ byggs upp och lagras i hjärnan är det troligt att dessa kandidater testades för sent i sjukdomen, under en fas då Aβ redan hade spelat det mesta av sin patogena roll. Nya kliniska studier i området där man gått in tidigare i sjukdomen med Aβ-riktade läkemedel har kunnat påvisa kliniska effekter i patient vilket stärker validiteten i denna målmekanism.
Att Aβ-amyloidos är sjukdomsorsakande i ärftlig, familjär Alzheimers är tydligt, som beskrivits ovan. Fler och fler jämförande studier, där Aβ processen i sporadisk Alzheimers jämförts med familjär Alzheimers, visar på en liknande uppbyggnad av Aβ i sporadisk sjukdom men att den oftast sker senare i livet. Dessa forskningsdata tyder starkt på att Aβ inlagring även spelar en helt avgörande patologisk roll i sporadisk Alzheimers, som utgör den stora gruppen, ca 99 procent, inom Alzheimers sjukdom.
Med tanke på dess nyckelroll i produktionen av toxiskt Aβ, är gammasekretas komplexet en mycket intressant terapeutisk målmekanism för potentiell behandling av Alzheimers sjukdom, och olika strategier för att interagera med detta enzymkomplex har undersökts:
Gamma sekretas hämmare (GSI):
GSI är läkemedel som inhiberar aktiviteten hos gamma sekretas. Genom att hämma gamma sekretas förhindrar GSI bildandet av Aβ42. Gamma sekretas har dock andra viktiga funktioner i kroppen, inklusive klyvningen av andra proteiner involverade i viktiga cellulära processer. Då GSI även hämmar processningen av dessa viktiga proteiner, kan detta leda till potentiella biverkningar. Hämningen av gamma sekretas orsakar bl.a. problem vid bearbetningen av Notch proteinet, som är avgörande för normal cellutveckling och differentiering. Därför har GSI:er haft problem i klinisk utveckling på grund av deras negativa sidoeffekter.
Gammasekretas modulatorer (GSM):
GSM:er, å andra sidan, inhiberar inte aktiviteten av gammasekretas utan modulerar dess funktion på ett mer selektivt sätt. De riktar sig specifikt in på klyvningen av APP via gamma sekretas utan att påverka andra substrat, såsom Notch. GSM modulerar specifikt hur gamma sekretas klyver ut Ab, så att det sker en minskning av produktionen av längre, aggregationsbenägna former av Aβ (såsom Aβ42) och en ökning av kortare former, som Aβ37 och 38, vilka inte är aggregationsbenägna och även har föreslagits ha skyddande effekter.
Fördelen med GSM framför GSI är deras selektivitet, vilket minskar risken för biverkningar som är associerade med en komplett inhibering av gamma sekretas aktivitet. Genom att modulera gamma sekretas klyvning mot att generera kortare former av Aβ, kan GSM:er minska bildningen av amyloida plack samtidigt som den normala fysiologiska funktionen av gamma sekretas bibehålls.
Läkemedelskandidaterna i Alzstatin-plattformen är GSM:er och har visat sig kunna minska produktionen av toxiskt Aβ42 med mer än 50 procent in vivo, utan att påverka andra signalvägar som är viktiga för celler. Dessutom ökas produktionen av kortare former av Aβ-peptiden, Aβ37 och Aβ38, som förutom att de inte är klibbiga och inte bildar aggregat, också kan hämma aggregeringen av toxiskt Aβ42. Prekliniska och kliniska data indikerar även på att de kan ha en potentiellt skyddande effekt. Detta innebär att läkemedelskandidaterna i Alzstatin-plattformen har två separata men samverkande effekter som tillsammans kan bidra till en starkare anti-amyloidogen och därför mer potent sjukdomsmodifierande effekt.
Vidare styrks projektet av positiva fynd gjorda i nyligen publicerade kliniska patientstudier med antikroppar såsom Aducanumab, Donanemab, och Lecanemab som vi bedömer validerar amyloidhypotesen som en behandlingsbar och kliniskt relevant patologisk mekanism. På CTAD konferensen i oktober 2023 visade Roche dessutom upp kliniska fas I-data med sin GSM, och kunde visa PoM i människa samt en god säkerhetsprofil för denna klass av substanser. De har nu gått in i fas II-studier under 2024, något som validerar denna målmekanism ytterligare och hjälper till att bana en regulatorisk väg framåt för denna substansklass. Stora framsteg har även tagits inom diagnostikfältet med nya blodbaserade tester, vilket medför en kostnadseffektiv möjlighet att screena högriskpopulationer, och därmed identifiera rätt patienter i sjukdomens presymptomatiska fas till kommande kliniska studier och framtida behandlingar.
ACD680 är läkemedelskandidaten inom Alzstatin och befinner sig för närvarande i preklinisk utvecklingsfas. ACD680 är en så kallad småmolekylär läkemedelskandidat vilket ger ett antal fördelar, bland annat möjliggör det oral administrering (tabletter), låga produktionskostnader och god penetration av blod-hjärnbarriären.
Du hittar våra Alzstatin publikationer: HÄR
För ytterligare information och material: se här
Filmen nedan visar översiktligt hur Alzstatin fungerar i hjärnan.